Unfiltered human PBMCs (10X Genomics)
Aedin Culhane, Michael Lynch, Maria Doyle
Dec 2nd, 2022
Source:vignettes/tenx-unfiltered-pbmc4k.Rmd
tenx-unfiltered-pbmc4k.RmdIntroduction
Here, we describe a brief analysis of the peripheral blood mononuclear cell (PBMC) dataset from 10X Genomics [@zheng2017massively]. The data are publicly available from the 10X Genomics website, from which we download the raw gene/barcode count matrices, i.e., before cell calling from the CellRanger pipeline.
This tutorial is edited and extracted from the OSCA Unfiltered human PBMCs workflow tenx.unfiltered-pbmc4k.Rmd
Data loading
library(DropletTestFiles)
raw.path <- getTestFile("tenx-2.1.0-pbmc4k/1.0.0/raw.tar.gz")
out.path <- file.path(tempdir(), "pbmc4k")
untar(raw.path, exdir=out.path)
library(DropletUtils)
fname <- file.path(out.path, "raw_gene_bc_matrices/GRCh38")
sce.pbmc <- read10xCounts(fname, col.names=TRUE)
library(scater)
rownames(sce.pbmc) <- uniquifyFeatureNames(
rowData(sce.pbmc)$ID, rowData(sce.pbmc)$Symbol)
library(EnsDb.Hsapiens.v86)
location <- mapIds(EnsDb.Hsapiens.v86, keys=rowData(sce.pbmc)$ID,
column="SEQNAME", keytype="GENEID")Quality control
We perform cell detection using the emptyDrops()
algorithm, as discussed in the book Advanced
Single-Cell Analysis with Bioconductor.
set.seed(100)
e.out <- emptyDrops(counts(sce.pbmc))
sce.pbmc <- sce.pbmc[,which(e.out$FDR <= 0.001)]
unfiltered <- sce.pbmcWe use a relaxed QC strategy and only remove cells with large mitochondrial proportions, using it as a proxy for cell damage. This reduces the risk of removing cell types with low RNA content, especially in a heterogeneous PBMC population with many different cell types.
stats <- perCellQCMetrics(sce.pbmc, subsets=list(Mito=which(location=="MT")))
high.mito <- isOutlier(stats$subsets_Mito_percent, type="higher")
sce.pbmc <- sce.pbmc[,!high.mito]
summary(high.mito)## Mode FALSE TRUE
## logical 3985 315
colData(unfiltered) <- cbind(colData(unfiltered), stats)
unfiltered$discard <- high.mito
gridExtra::grid.arrange(
plotColData(unfiltered, y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, y="subsets_Mito_percent",
colour_by="discard") + ggtitle("Mito percent"),
ncol=2
)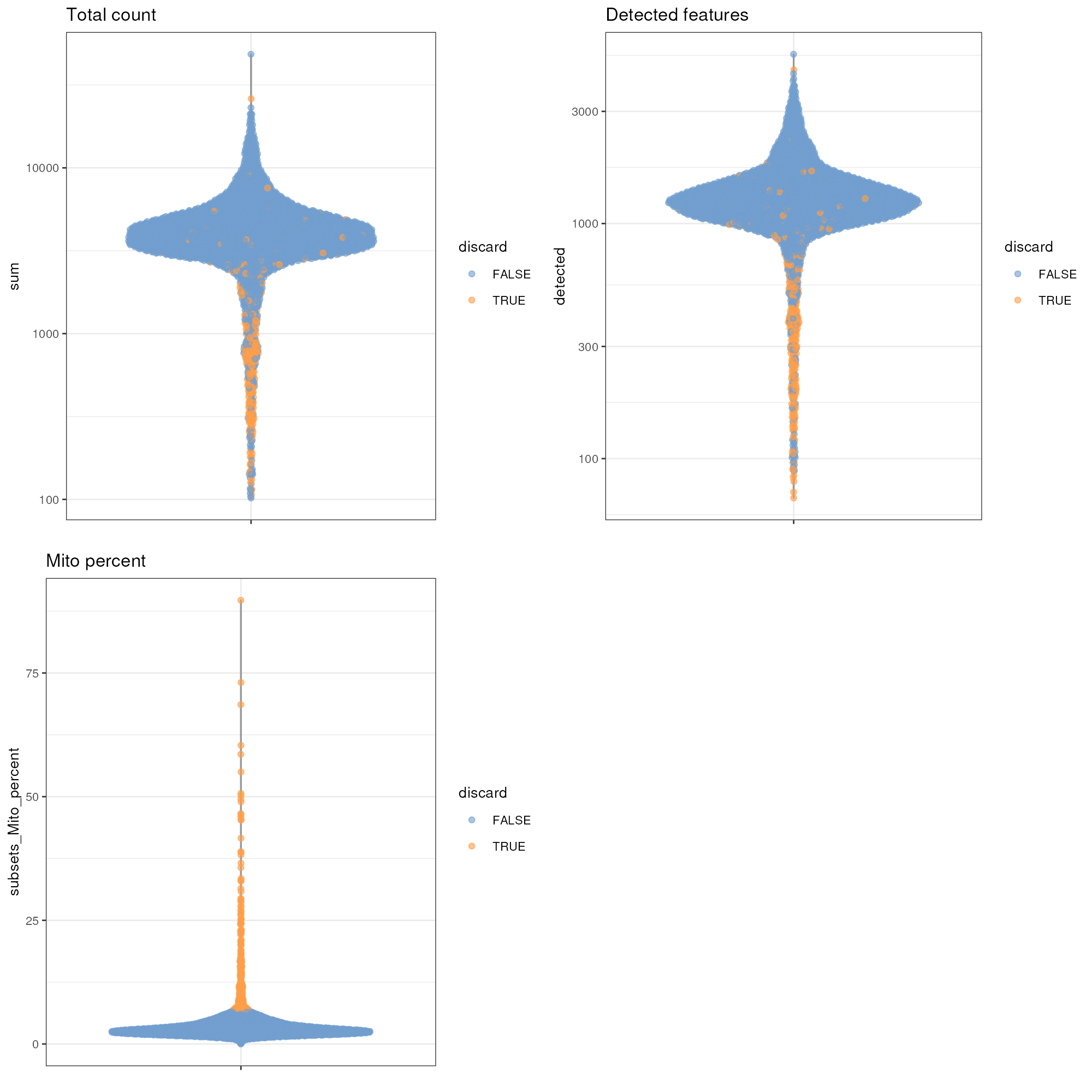
Distribution of various QC metrics in the PBMC dataset after cell calling. Each point is a cell and is colored according to whether it was discarded by the mitochondrial filter.
plotColData(unfiltered, x="sum", y="subsets_Mito_percent",
colour_by="discard") + scale_x_log10()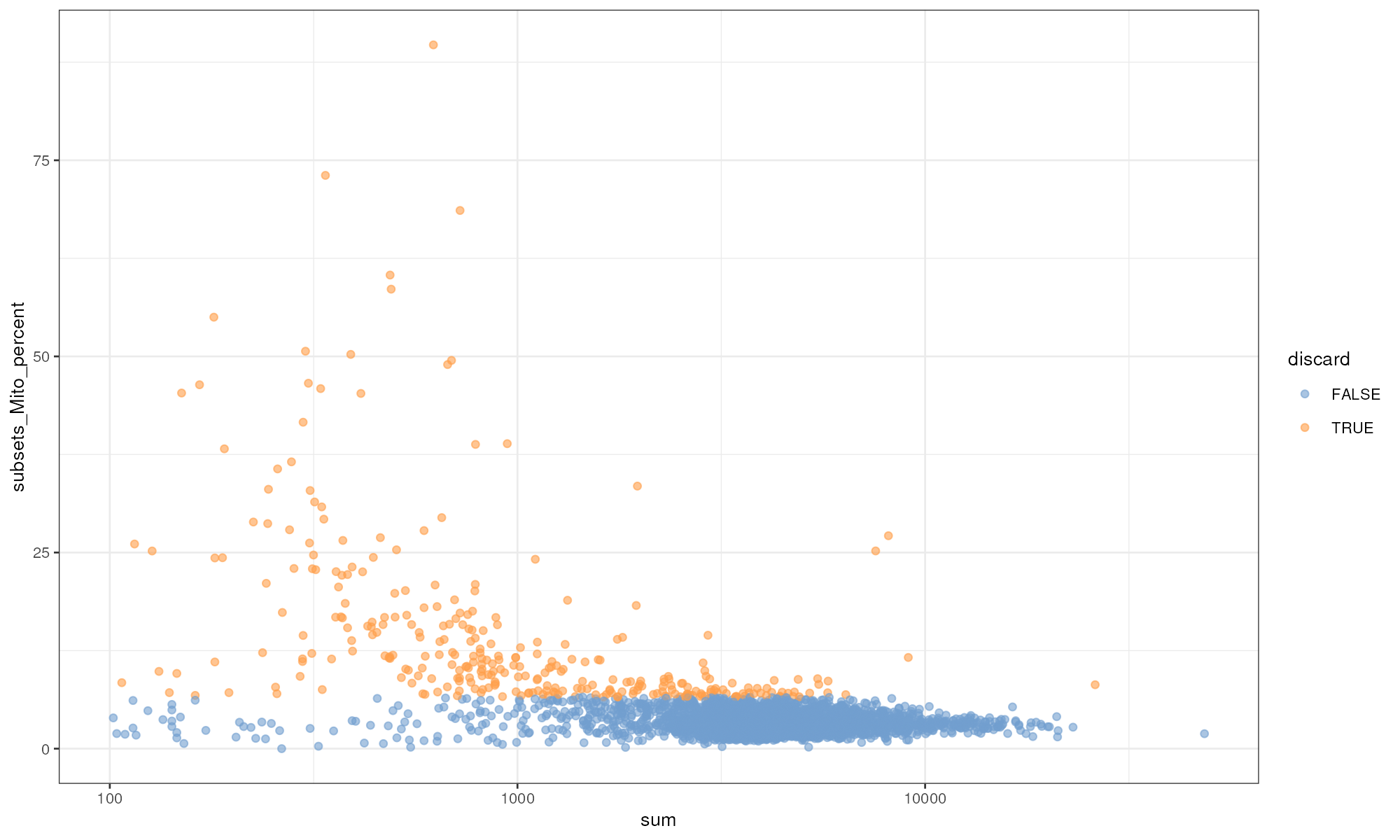
Proportion of mitochondrial reads in each cell of the PBMC dataset compared to its total count.
Normalization
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.pbmc)
sce.pbmc <- computeSumFactors(sce.pbmc, cluster=clusters)
sce.pbmc <- logNormCounts(sce.pbmc)
summary(sizeFactors(sce.pbmc))## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.00749 0.71207 0.87490 1.00000 1.09900 12.25412
plot(librarySizeFactors(sce.pbmc), sizeFactors(sce.pbmc), pch=16,
xlab="Library size factors", ylab="Deconvolution factors", log="xy")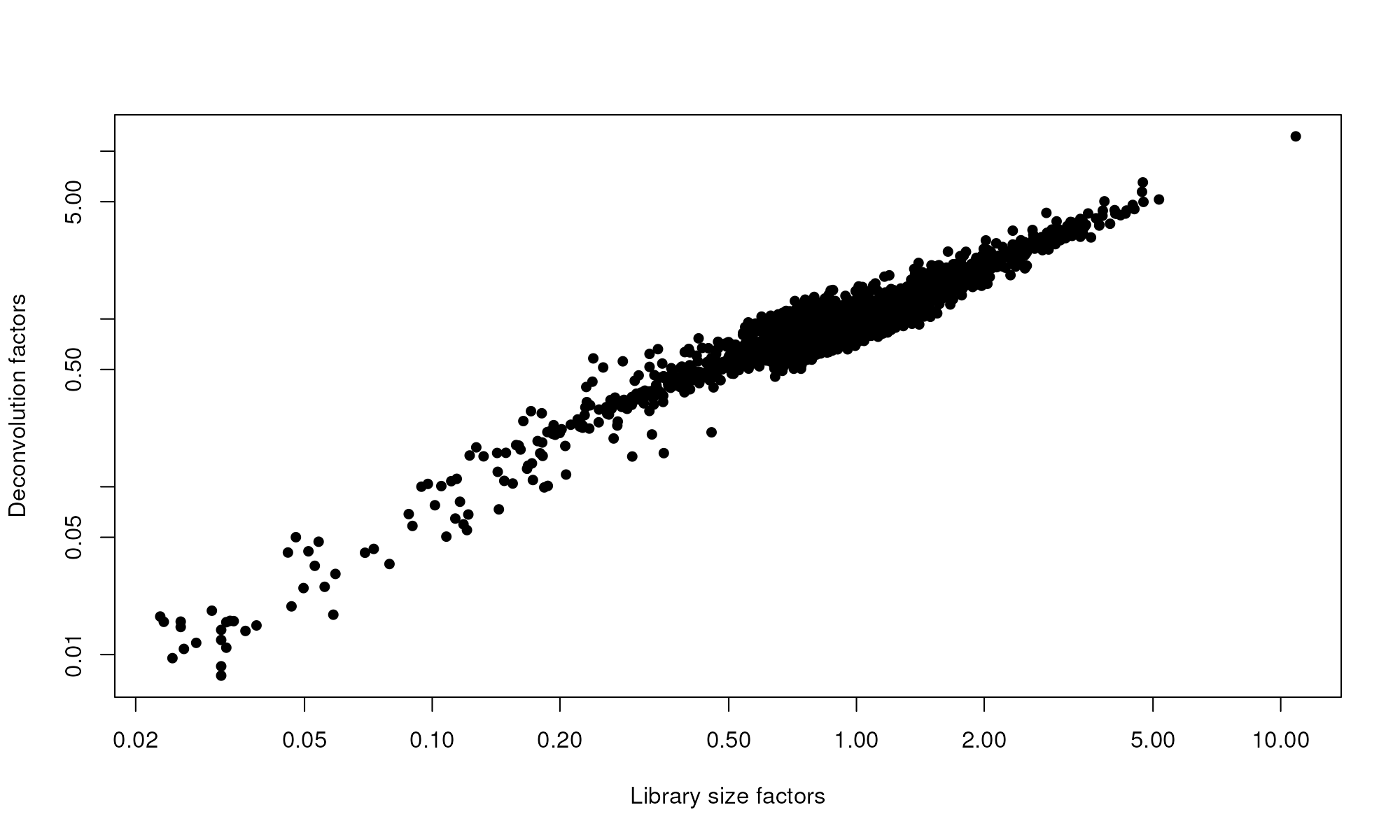
Relationship between the library size factors and the deconvolution size factors in the PBMC dataset.
Variance modelling
set.seed(1001)
dec.pbmc <- modelGeneVarByPoisson(sce.pbmc)
top.pbmc <- getTopHVGs(dec.pbmc, prop=0.1)
plot(dec.pbmc$mean, dec.pbmc$total, pch=16, cex=0.5,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(dec.pbmc)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)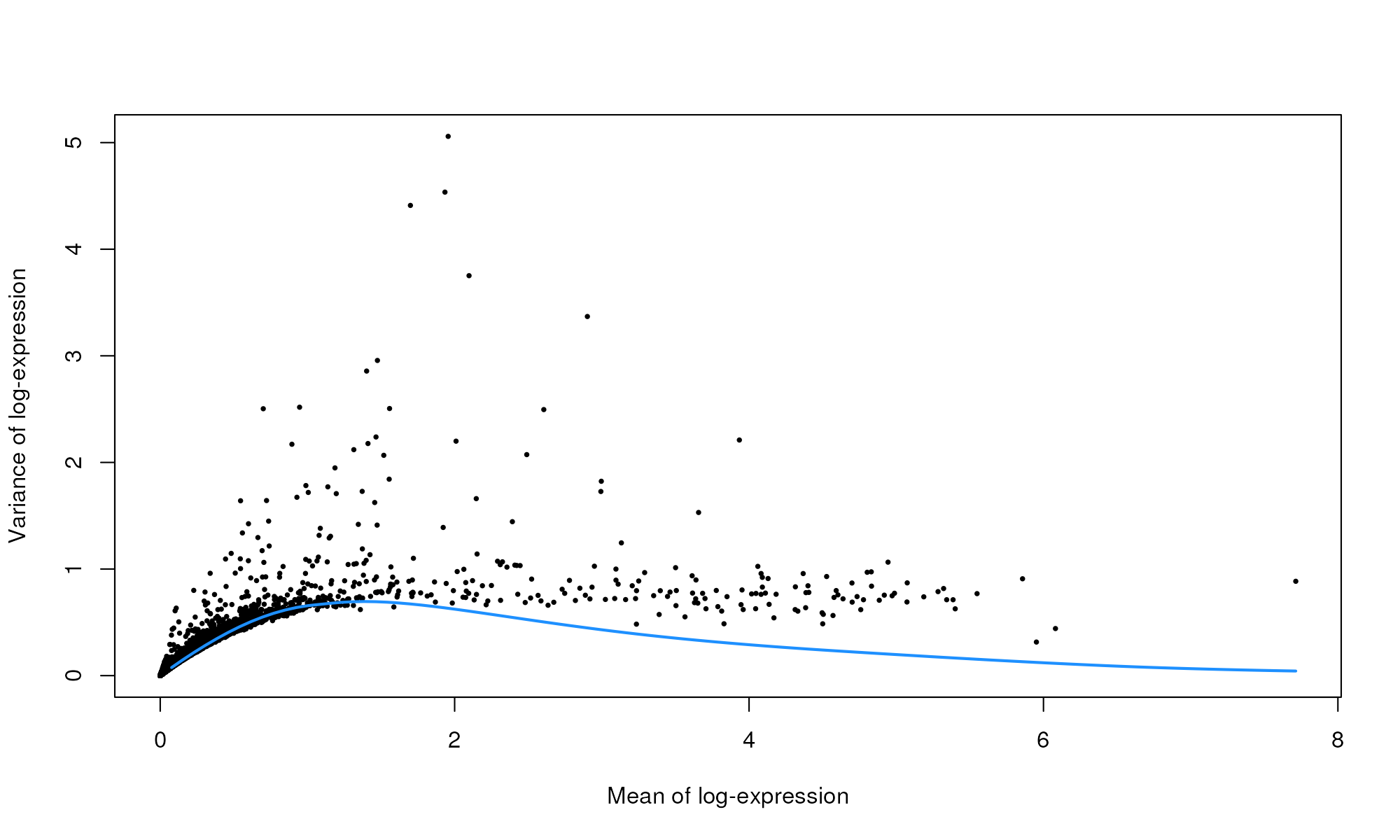
Per-gene variance as a function of the mean for the log-expression values in the PBMC dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to simulated Poisson counts.
Dimensionality reduction
set.seed(10000)
sce.pbmc <- denoisePCA(sce.pbmc, subset.row=top.pbmc, technical=dec.pbmc)
set.seed(100000)
sce.pbmc <- runTSNE(sce.pbmc, dimred="PCA")
set.seed(1000000)
sce.pbmc <- runUMAP(sce.pbmc, dimred="PCA")We verify that a reasonable number of PCs is retained.
ncol(reducedDim(sce.pbmc, "PCA"))## [1] 9Clustering
g <- buildSNNGraph(sce.pbmc, k=10, use.dimred = 'PCA')
clust <- igraph::cluster_walktrap(g)$membership
colLabels(sce.pbmc) <- factor(clust)##
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
## 205 508 541 56 374 125 46 432 302 867 47 155 166 61 84 16
plotTSNE(sce.pbmc, colour_by="label")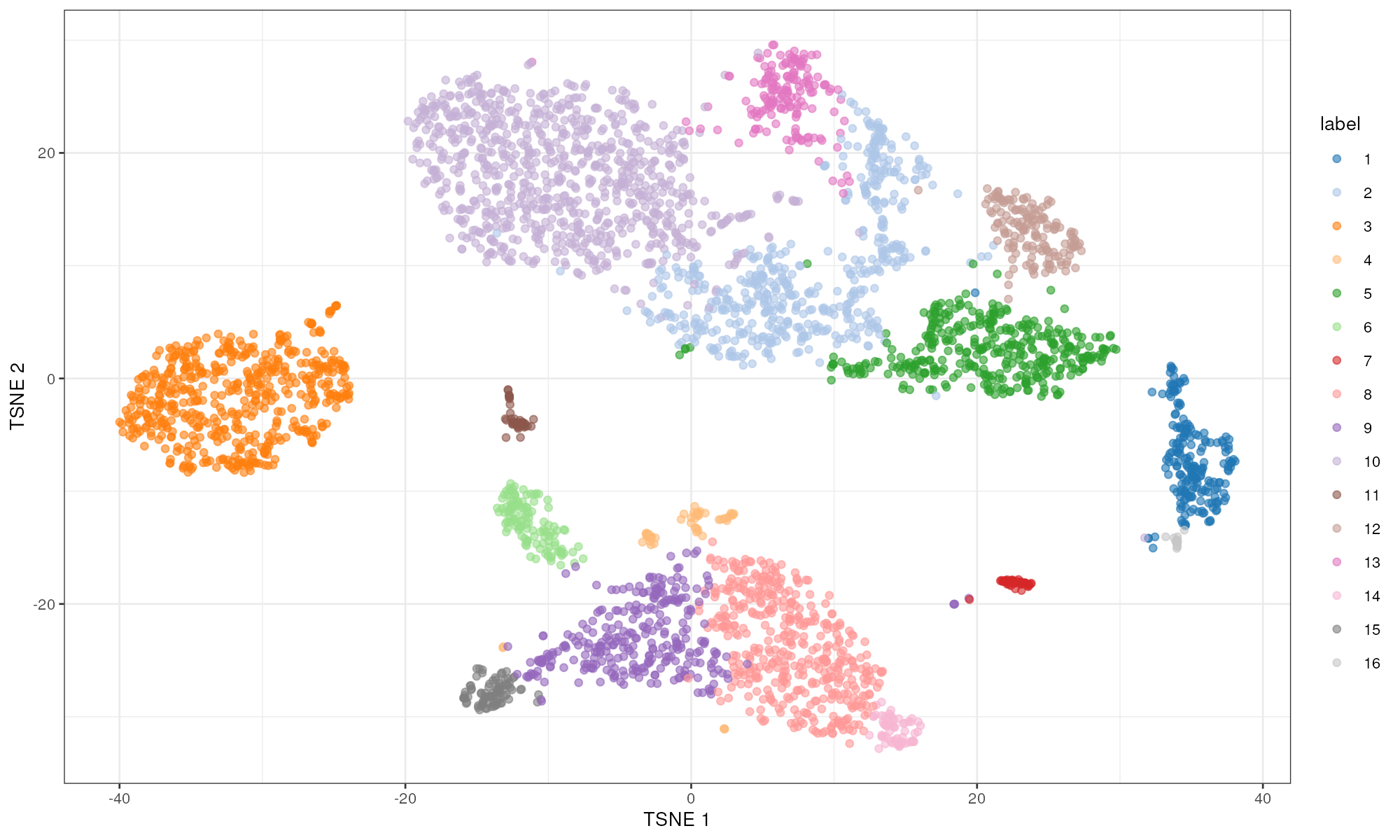
Obligatory \(t\)-SNE plot of the PBMC dataset, where each point represents a cell and is colored according to the assigned cluster.
Interpretation
markers <- findMarkers(sce.pbmc, pval.type="some", direction="up")We examine the markers for cluster 8 in more detail. High expression of CD14, CD68 and MNDA combined with low expression of CD16 suggests that this cluster contains monocytes, compared to macrophages in cluster 15 (Figure @ref(fig:unref-mono-pbmc-markers)).
marker.set <- markers[["8"]]
as.data.frame(marker.set[1:30,1:3])## p.value FDR summary.logFC
## CSTA 7.170624e-222 2.015964e-217 2.4178954
## MNDA 1.196631e-221 2.015964e-217 2.6614935
## FCN1 2.375980e-213 2.668543e-209 2.6380934
## S100A12 4.393470e-212 3.700839e-208 3.0808902
## VCAN 1.711043e-199 1.153038e-195 2.2603760
## TYMP 1.173532e-154 6.590164e-151 2.0237930
## AIF1 3.673649e-149 1.768285e-145 2.4603604
## LGALS2 4.004740e-137 1.686696e-133 1.8927606
## MS4A6A 5.639909e-134 2.111457e-130 1.5457061
## FGL2 2.044513e-124 6.888781e-121 1.3859366
## RP11-1143G9.4 6.891551e-122 2.110945e-118 2.8042347
## AP1S2 1.786019e-112 5.014842e-109 1.7703547
## CD14 1.195352e-110 3.098169e-107 1.4259764
## CFD 6.870490e-109 1.653531e-105 1.3560255
## GPX1 9.048825e-107 2.032607e-103 2.4013937
## TNFSF13B 3.920319e-95 8.255701e-92 1.1151275
## KLF4 3.309726e-94 6.559876e-91 1.2049050
## GRN 4.801206e-91 8.987324e-88 1.3814668
## NAMPT 2.489624e-90 4.415020e-87 1.1438687
## CLEC7A 7.736088e-88 1.303299e-84 1.0616120
## S100A8 3.124930e-84 5.013875e-81 4.8051993
## SERPINA1 1.580359e-82 2.420392e-79 1.3842689
## CD36 8.018347e-79 1.174653e-75 1.0538169
## MPEG1 8.481588e-79 1.190744e-75 0.9778095
## CD68 5.118714e-78 6.898798e-75 0.9481203
## CYBB 1.200516e-77 1.555776e-74 1.0300245
## S100A11 1.174556e-72 1.465759e-69 1.8962486
## RBP7 2.467027e-71 2.968714e-68 0.9666127
## BLVRB 3.762610e-71 4.371634e-68 0.9701168
## CD302 9.859086e-71 1.107307e-67 0.8792077
plotExpression(sce.pbmc, features=c("CD14", "CD68",
"MNDA", "FCGR3A"), x="label", colour_by="label")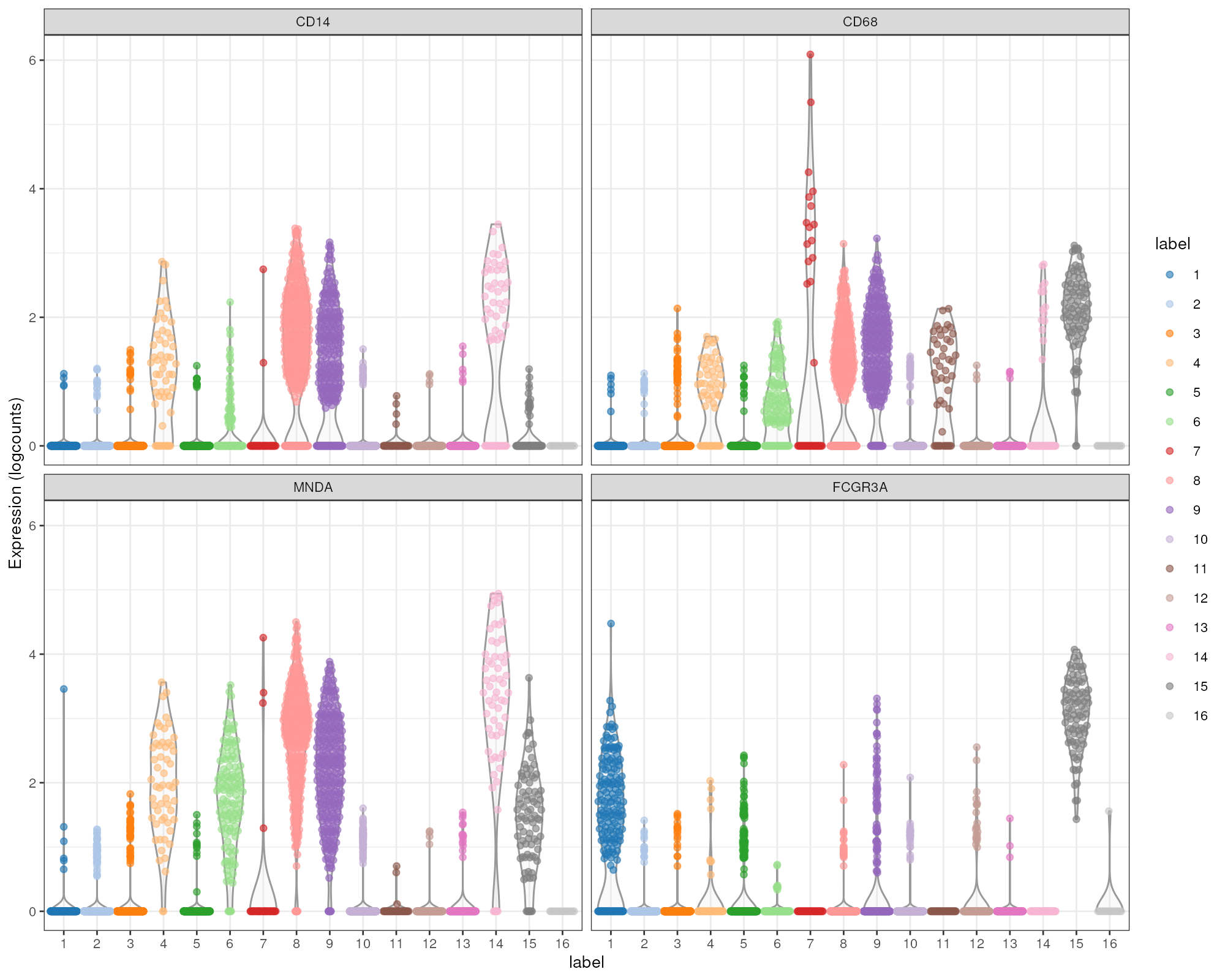
Distribution of expression values for monocyte and macrophage markers across clusters in the PBMC dataset.
Session Info
R version 4.2.2 (2022-10-31) Platform: x86_64-pc-linux-gnu (64-bit) Running under: Ubuntu 22.04.1 LTS
Matrix products: default BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3 LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so
locale: [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages: [1] stats4 stats graphics grDevices utils
datasets methods
[8] base
other attached packages: [1] scran_1.26.0
EnsDb.Hsapiens.v86_2.99.0
[3] ensembldb_2.22.0 AnnotationFilter_1.22.0
[5] GenomicFeatures_1.50.2 AnnotationDbi_1.60.0
[7] scater_1.26.1 ggplot2_3.4.0
[9] scuttle_1.8.1 DropletUtils_1.18.1
[11] SingleCellExperiment_1.20.0 SummarizedExperiment_1.28.0 [13]
Biobase_2.58.0 GenomicRanges_1.50.1
[15] GenomeInfoDb_1.34.4 IRanges_2.32.0
[17] S4Vectors_0.36.0 BiocGenerics_0.44.0
[19] MatrixGenerics_1.10.0 matrixStats_0.63.0
[21] DropletTestFiles_1.8.0 knitr_1.41
loaded via a namespace (and not attached): [1] AnnotationHub_3.6.0
BiocFileCache_2.6.0
[3] systemfonts_1.0.4 igraph_1.3.5
[5] lazyeval_0.2.2 BiocParallel_1.32.4
[7] digest_0.6.30 htmltools_0.5.3
[9] viridis_0.6.2 fansi_1.0.3
[11] magrittr_2.0.3 memoise_2.0.1
[13] ScaledMatrix_1.6.0 cluster_2.1.4
[15] limma_3.54.0 Biostrings_2.66.0
[17] R.utils_2.12.2 pkgdown_2.0.6
[19] prettyunits_1.1.1 colorspace_2.0-3
[21] blob_1.2.3 rappdirs_0.3.3
[23] ggrepel_0.9.2 textshaping_0.3.6
[25] xfun_0.35 dplyr_1.0.10
[27] crayon_1.5.2 RCurl_1.98-1.9
[29] jsonlite_1.8.3 glue_1.6.2
[31] gtable_0.3.1 zlibbioc_1.44.0
[33] XVector_0.38.0 DelayedArray_0.24.0
[35] BiocSingular_1.14.0 Rhdf5lib_1.20.0
[37] HDF5Array_1.26.0 scales_1.2.1
[39] DBI_1.1.3 edgeR_3.40.0
[41] Rcpp_1.0.9 viridisLite_0.4.1
[43] xtable_1.8-4 progress_1.2.2
[45] dqrng_0.3.0 bit_4.0.5
[47] rsvd_1.0.5 metapod_1.6.0
[49] httr_1.4.4 FNN_1.1.3.1
[51] ellipsis_0.3.2 farver_2.1.1
[53] pkgconfig_2.0.3 XML_3.99-0.12
[55] R.methodsS3_1.8.2 uwot_0.1.14
[57] sass_0.4.4 dbplyr_2.2.1
[59] locfit_1.5-9.6 utf8_1.2.2
[61] labeling_0.4.2 tidyselect_1.2.0
[63] rlang_1.0.6 later_1.3.0
[65] munsell_0.5.0 BiocVersion_3.16.0
[67] tools_4.2.2 cachem_1.0.6
[69] cli_3.4.1 generics_0.1.3
[71] RSQLite_2.2.19 ExperimentHub_2.6.0
[73] evaluate_0.18 stringr_1.4.1
[75] fastmap_1.1.0 yaml_2.3.6
[77] ragg_1.2.4 bit64_4.0.5
[79] fs_1.5.2 purrr_0.3.5
[81] KEGGREST_1.38.0 sparseMatrixStats_1.10.0
[83] mime_0.12 R.oo_1.25.0
[85] xml2_1.3.3 biomaRt_2.54.0
[87] compiler_4.2.2 beeswarm_0.4.0
[89] filelock_1.0.2 curl_4.3.3
[91] png_0.1-8 interactiveDisplayBase_1.36.0 [93] statmod_1.4.37
tibble_3.1.8
[95] bslib_0.4.1 stringi_1.7.8
[97] highr_0.9 desc_1.4.2
[99] bluster_1.8.0 lattice_0.20-45
[101] ProtGenerics_1.30.0 Matrix_1.5-3
[103] vctrs_0.5.1 pillar_1.8.1
[105] lifecycle_1.0.3 rhdf5filters_1.10.0
[107] BiocManager_1.30.19 jquerylib_0.1.4
[109] BiocNeighbors_1.16.0 bitops_1.0-7
[111] irlba_2.3.5.1 httpuv_1.6.6
[113] rtracklayer_1.58.0 R6_2.5.1
[115] BiocIO_1.8.0 promises_1.2.0.1
[117] gridExtra_2.3 vipor_0.4.5
[119] codetools_0.2-18 assertthat_0.2.1
[121] rhdf5_2.42.0 rprojroot_2.0.3
[123] rjson_0.2.21 withr_2.5.0
[125] GenomicAlignments_1.34.0 Rsamtools_2.14.0
[127] GenomeInfoDbData_1.2.9 parallel_4.2.2
[129] hms_1.1.2 grid_4.2.2
[131] beachmat_2.14.0 rmarkdown_2.18
[133] DelayedMatrixStats_1.20.0 Rtsne_0.16
[135] shiny_1.7.3 ggbeeswarm_0.6.0
[137] restfulr_0.0.15